 Abteilung Reinst- und Mikroproduktion
Abteilung Reinst- und Mikroproduktion
Reinheit von Medizinprodukten
Per Gesetz sind Medizintechnikunternehmen für ihre Produkte haftbar. Sie verpflichten sich dazu, bei bestimmungsgemäßer Anwendung ihrer Produkte die Risiken für die Patienten zu minimieren. In diesem Zusammenhang nehmen auch die Forderungen nach der Betrachtung der Reinheit von Medizinprodukten zu. Im Spannungsfeld zwischen Produkthaftung einerseits und der häufig unzureichenden Normung andererseits sind Medizintechnikunternehmen und deren Zulieferer auf sich selbst gestellt. Der eigenverantwortliche Umgang mit dem Thema Reinheit führte dazu, dass unterschiedlichste Ansätze zur Bewertung und Beurteilung der Reinheit eingesetzt wurden. Die dadurch entstehende Lücke schießt die neue VDI-Richtlinie 2083 Blatt 21 »Reinheit von Medizinprodukten im Herstellungsprozess«.
VDI-Richtlinie 2083 Blatt 21
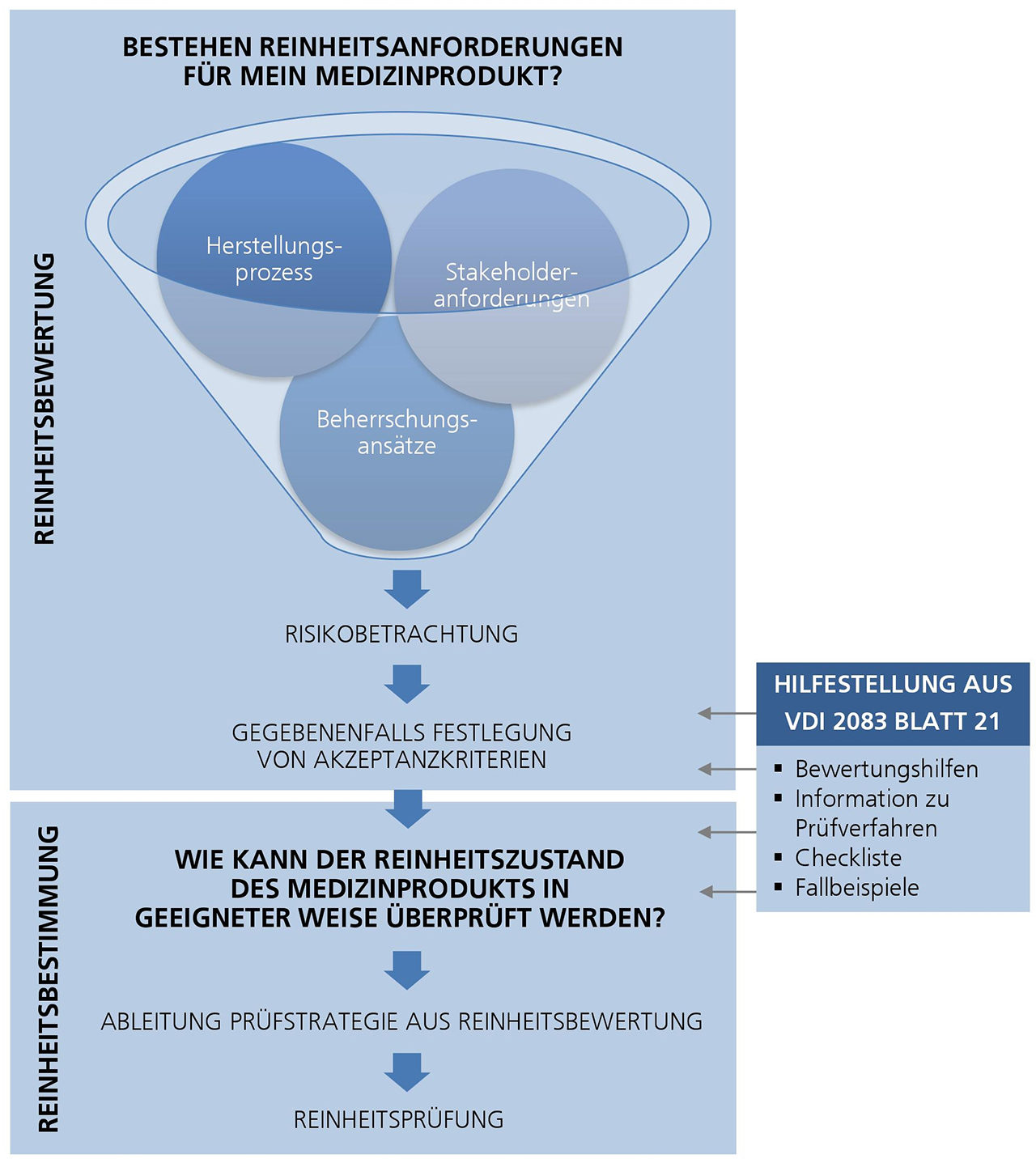
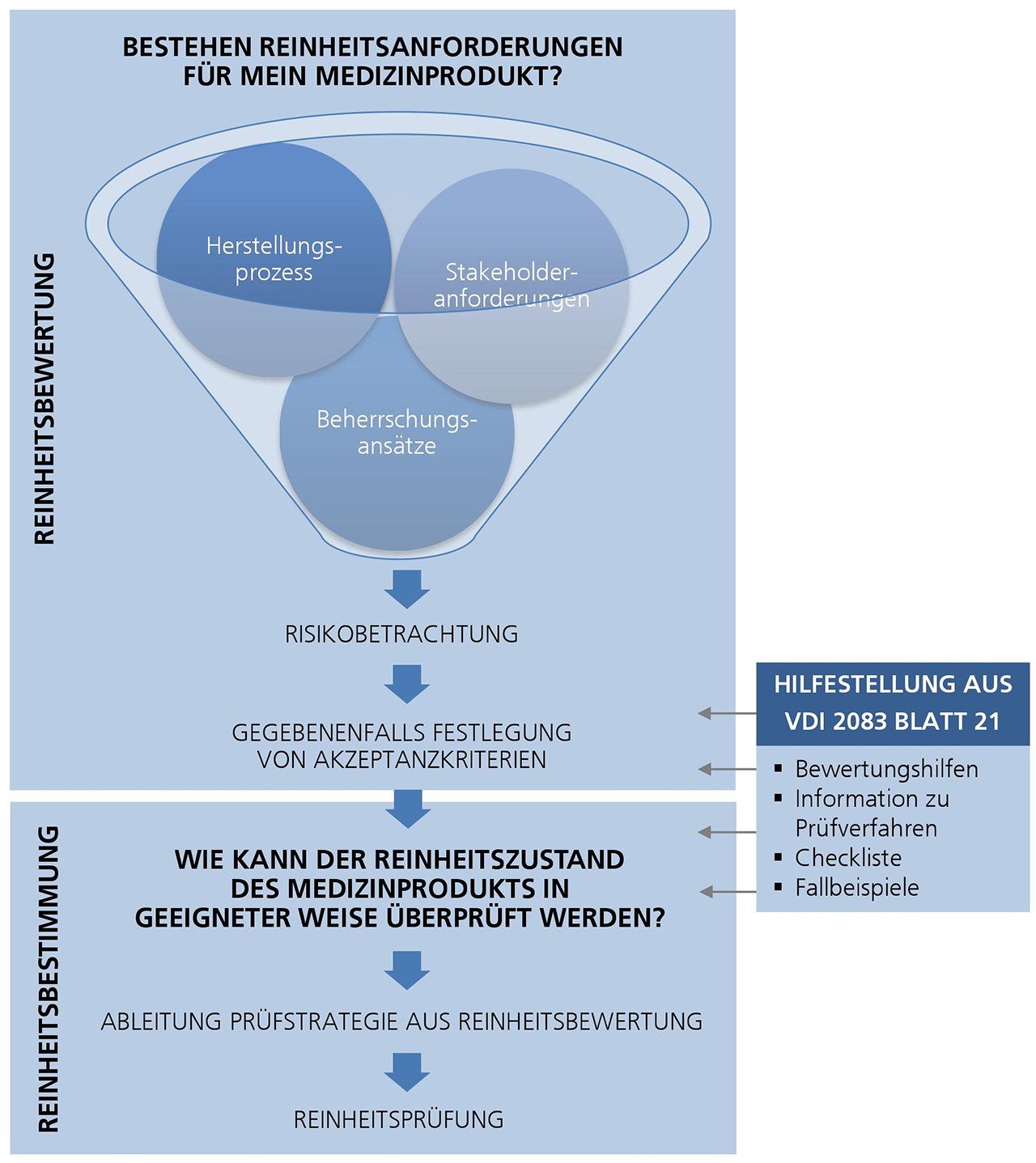
Die neue VDI-Richtlinie 2083 Blatt 21 ist ein allgemein anwendbarer und übergeordneter Standard zur Bewertung der Reinheit von Medizinprodukten im Herstellungsprozess und bildet somit eine Basis bei fehlender produktspezifischer Regelung.
Sie beinhaltet zum einen die Vorgehensweise für die Festlegung eines geeigneten Prüfverfahrens. Zum anderen werden Hilfestellungen zur strukturellen Vorgehensweise für die Ableitung von Akzeptanzkriterien für kritische Verunreinigungen, auf Basis eines risikobasierten Ansatzes, gegeben. Fallbeispiele verdeutlichen die Thematik.
Noch beantwortet die neue VDI-Richtlinie 2083 Blatt 21 nicht alle Fragestellungen. Bei der produktspezifischen Umsetzung sind die Hersteller noch immer auf sich selbst gestellt. Je nach gewähltem Weg können bei vergleichbaren Produkten unterschiedliche Akzeptanzkriterien das Ergebnis sein. Um den Status Quo weiter zu verbessern, eignet sich ein Industrieverbund.
Industrieverbund ACCEPT-MED

Im Rahmen des neuen Industrieverbunds »Reinheit von Medizinprodukten« arbeiten die betroffenen Beteiligten eng zusammenarbeiten. Das Ziel des Industrieverbunds ist die Erarbeitung einer Strategie zur Ableitung von Akzeptanzkriterien aufgrund medizinischer Aspekte sowie die Ermittlung des fertigungstechnisch herstellbaren Ist-Zustandes.

 Am 26. November startete am Fraunhofer IPA der Industrieverband Reinheit von Medizinprodukten im Herstellungsprozess nach VDI 2036 Blatt 21.
Am 26. November startete am Fraunhofer IPA der Industrieverband Reinheit von Medizinprodukten im Herstellungsprozess nach VDI 2036 Blatt 21.Der Verbund bietet eine Kommunikations-Plattform und ermöglicht einen fachlichen und offenen Wissensaustausch im Kreis von Medizintechnikproduzenten. Projektstart ist der 21. April 2020.
Das Ergebnis soll eine Handlungshilfe sein, die die Ableitung produktspezifischer Akzeptanzkriterien ermöglicht. Das vereinfacht den Dialog mit den Benannten Stellen, sowie die Diskussion mit Zulieferern und Dienstleistern. Um die Ableitung von Akzeptanzkriterien für produktspezifische Reinheitsanforderungen weiter zu konkretisieren, soll der in der VDI 2083 Blatt 21 beschriebene Ansatz, der den strukturellen Weg nach aktuellen Stand der Technik beschreibt, weiter für partikuläre und chemische Verunreinigungen konkretisiert werden.
Leistungsangebot
Die Abteilung Reinst- und Mikroproduktion besitzt ein umfangreiches Leistungsangebot zur Bewertung und Bestimmung der Reinheit von Medizinprodukten:
- Reinigung & Verpackung
- Reinigungsvalidierung
- Analyse partikulärer, chemischer & mikrobiologischer Verunreinigungen zum Beispiel zur Bestimmung der Produktreinheit
- Ursachenforschung
- Planung und Optimierung von Fertigungsumgebungen
- Beratung und Schulung


In-vitro-Zytotoxizität
Alle Medizinprodukte, die in Verkehr gebracht werden, müssen vor ihrer Zulassung zahlreiche Prüfungen durchlaufen, um das Risiko für Patienten, Anwender und Dritte auf ein Minimum zu reduzieren. Je nach Klassifizierung des Medizinproduktes sind verschiedene Prüfungen der Normenreihe ISO 10993 durchzuführen.
Im Rahmen dieser notwendigen Prüfungen ist die Beurteilung der In-vitro-Zytotoxizität, die Hinweise zum Reinheitszustand und der Biokompatibilität (Körperverträglichkeit) liefert, die wichtigste initiale Untersuchung.
Für die Prüfung der In-vitro-Zytotoxizität nach DIN EN ISO 10993-5 verwenden wir die in den meisten Prüflaboren eingesetzte Mauszelllinie L929. Daneben bieten wir die Prüfung auch an der menschlichen Hautzelllinie HaCaT an, um die Anwendungssituation der zu prüfenden Produkte (je nach Anwendung am od. im Menschen) besser zu berücksichtigen.
Neben der biologischen Beurteilung von Medizinprodukten können auch Materialien aus Forschung und Entwicklung sowie anderen Industriebranchen auf ihre In-vitro-Zytotoxizität überprüft werden, um sie zukünftig als Komponente eines Medizinproduktes einsetzen zu können. Der Test detektiert auch mögliche Verunreinigungen wie z. B. Rückstände von Reinigungslösungen oder anderen Hilfsmitteln, die aus der Produktion auf dem Produkt zurückbleiben. In Kombination mit weiteren Prüfverfahren lassen sich diese Kontaminationen zuverlässig identifizieren.
Qualitätsmanagement und Verifizierung
Durch das Qualitätsmanagementsystem nach DIN EN ISO/IEC 17025:2018 ist gewährleistet, dass die Prüfungen von kompetentem, qualifiziertem Personal mit metrologisch rückgeführter Geräteausstattung durchgeführt werden.
Akkreditierte Prüfverfahren
Unsere Prüfverfahren wurden von der Deutschen Akkreditierungsstelle DAkkS sowie der Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten ZLG begutachtet. Seit Oktober 2022 ist unser Prüflabor für die genannten Prüfungen akkreditiert:
- Bestimmung der Toxizität von Forschungs- und Industrieprodukten mittels biologischer Prüfverfahren (DIN EN ISO 10993-5): Biologische Beurteilung von Medizinprodukten - Teil 5: Prüfungen auf In-vitro-Zytotoxizität unter Verwendung von L929 / HaCaT-Zellen (hier: abweichend nicht zur Konformitätsbewertung für Medizinprodukte) (D-PL-11140-07-01) (DAkkS)
- Biologische Beurteilung von Medizinprodukten - Teil 5: Prüfungen auf In-vitro-Zytotoxizität (DIN EN ISO 10993-5) (D-PL-11140-07-02) (DAkkS/ZLG)
- Prüflaboratorium (ipa.fraunhofer.de)
Reinheit von Medizinprodukten
Reinheitsprüfung für partikuläre Verunreinigungen am IPA
